REVIEW
A contemporary review of the diagnosis and management of vascular Ehlers–Danlos syndrome
Chen A,1 Stacey C, Coughlin P2
Plain English Summary
Why we undertook the work: Vascular Ehlers–Danlos syndrome (vEDS) is a rare genetic condition (ie, that you are born with) that affects the strength of major blood vessels (arteries). This means that arteries are fragile and, as such, are prone to damage with even minimal or no trauma.
What we did: We reviewed the literature searching for papers which focused on how we may diagnose and manage patients with vEDS.
What we found: Diagnosis is still based on recognised clinical features, but there is some hope that objective tests could help with the diagnosis. The major aim of treatment is to prevent blood vessel complications, and the evidence would point to the use of a drug that affects the pressure of blood as it passes through the blood vessels. When complications do happen they are difficult to treat, so the aim should be to reduce the chance of such complications occurring.
What this means: Early diagnosis and active prevention of complications are key in the management of patients with vEDS.
Abstract
Introduction: Vascular Ehlers–Danlos syndrome (vEDS) is a connective tissue disorder weakening the vasculature. There is a perceived lack of awareness within the medical fraternity on this condition. This paper reviews contemporary evidence to enhance the understanding of this complex condition.
Methods: A systematic review was performed. Inclusion criteria were studies in humans with a minimum of five patients. Exclusion criteria were laboratory or animal-based studies, reviews and studies not in English. A total of 115 papers were assessed with articles grouped into four categories: diagnosis, natural history, medical and surgery therapy.
Results: The Villefranche criteria have a high sensitivity/negative predictive value for diagnosing EDS. Assessment of skin architecture/radiological investigation of large vessels can facilitate diagnosis. Genetic testing is available for the pathogenic COL3A1 variant. With regard to natural history, an overall review of 1,400 patients showed that age at diagnosis ranged from 4 to 40 years, age of death ranged from 28 to 54 years and vascular complications were frequent. The key medical therapy is celiprolol, which is associated with a reduction in arterial events three times greater with treatment than without. Non-operative therapy/non-invasive imaging techniques are suggested to be used when possible. Vessel reconstruction is possible but fragile tissue makes this challenging and endovascular interventions are ideally suited for the treatment of false aneurysms. There is a high rate of procedure-related complications (early and late).
Conclusions: The poor natural history of vEDS means that accurate diagnosis is key to allow extension of complication-free periods. Vascular interventions are associated with high complication rates.
Introduction
Ehlers–Danlos syndrome (EDS) is a recognised connective tissue disorder that affects collagen and extracellular matrix function. EDS is a cluster of 13 inherited conditions/types (19 different causal genes) which vary in incidence, clinical presentation and natural history.1
The most common of these is that of hypermobile EDS which has an incidence of approximately 1:10,000 of the population.2 Vascular EDS (vEDS) or – as it was previously called – type IV is less common but is of interest to vascular surgeons due to the effect it has on the extracellular matrix of both the vasculature and also within hollow visceral organs. vEDS is dominantly inherited and is caused by a mutation in the gene COL3A1 which encodes the alpha 1 chain of type III collagen.1
Because of a clinical overlap with some forms of Loeys–Dietz syndrome, Marfan syndrome and familial arterial aneurysm and dissection syndromes, the diagnosis should be confirmed by identification of pathogenic variants in COL3A1 to allow for appropriate surveillance, treatment and family studies.3
Over the last four decades there has been progress in the understanding of vEDS. This has been driven in part by some understanding of the underlying pathological and molecular processes that underpin vEDS.1 Yet there is still a lack of awareness around the condition in the medical fraternity in general and in vascular surgeons specifically.
The aim of this review is to explore the current available information for the diagnosis, medical and surgery therapy and natural history for patients with vEDS to highlight this condition to vascular surgeons and allied healthcare professionals. This paper also provides a patient’s perspective on the condition to highlight barriers experienced in accessing appropriate care.
Methods
A systematic review of published material relating to vEDS was undertaken. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (http://www.prisma-statement.org/) were adhered to throughout. Figure 1 shows the study flow chart.
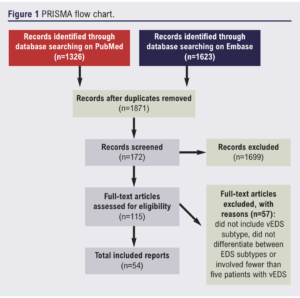
PubMed and Embase databases were queried, taking into account the different notations for the EDS subtypes using the following keywords: vascular, type 4, type IV. Based on these keywords, we queried the PubMed database with the following search string: (vascular OR “type 4” OR “type IV”) AND (Ehlers Danlos OR Ehlers-Danlos). The Embase database was also queried for additional results with the following search string: (vascular OR type 4 OR type IV) AND (Ehlers Danlos OR Ehlers-Danlos). All material published before 17 April 2021 were eligible for inclusion in this review.
After removing duplicate results, studies were included if they involved five or more patients with the vEDS subtype. Studies that did not include the vEDS subtype or did not differentiate between EDS subtypes were excluded. The following types of studies were considered: clinical trials, case–control studies, cross-sectional studies, cohort studies, case series and case reports. We excluded cell culture laboratory studies, animal studies, reviews and studies that were not published in English. A bibliography was created using Zotero (https://www.zotero.org/). Using these criteria, two authors (AC and PC) screened the titles and abstracts of the remaining papers and removed those that did not meet the inclusion criteria. Further papers from the bibliography were reviewed when appropriate.
Results
A total of 115 papers were assessed for eligibility and, after analysis, a total of 54 papers were assessed to formulate this review. Due to the nature of this narrative and clinical focused synthesis, we grouped the articles into diagnosis of vEDS, medical and surgery management and the natural history of vEDS. Papers that focused on the genetics of vEDS were not included in this review.
Diagnosis
The diagnosis of vEDS is challenging and patients often arrive at a diagnosis after a long and protracted path of misdiagnosis or uncertainty.
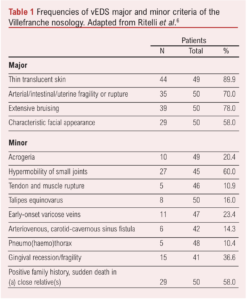
Initial diagnostic criteria for EDS overall were initially proposed in 1986. The progress in our understanding and the specific identification of a vascular specific EDS led to the development of the Villefranche criteria in 1997, which was put forward to improve patient selection for genetic testing.4 The Villefranche criteria consist of four major diagnostic criteria – arterial/intestinal/uterine fragility or rupture, extensive bruising, thin/translucent skin and characteristic facial appearance – with the presence of two or more criteria identifying those patients who would benefit from genetic testing.
Minor criteria include acrogeria (a skin condition characterised by premature aging with unusually fragile, thin skin on the hands and feet), small joint hypermobility, tendon/muscle rupture, clubfoot, early onset varicose veins, arteriovenous or carotid-cavernous sinus fistula, pneumothorax, gingival recession and a positive family history, sudden death in a close relative. The diagnostic relevance of these has been untested.
A study by Henneton et al retrospectively looked at a consecutive series of patients (384 probands, defined as the first person in a family to receive genetic counselling and/or testing for suspected hereditary risk and 135 relatives) with a possible diagnosis of vEDS in a tertiary referral unit who were scored using the major and minor diagnostic criteria of the Villefranche classification and also went on to be genetically tested.5 Of these patients, a specific pathogenic COL3A1 variant was confirmed in 165 patients but the Villefranche criteria for diagnosis was met in 248 patients (sensitivity 79%, negative predictive value (NPV) 87%). Diagnostic accuracy was highest for symptomatic probands (sensitivity 92%, NPV 95%). The sensitivity and NPV of the Villefranche criteria were lower for relatives yet, in practice, this group of patients is likely to undergo mandatory genetic testing and thus it could be argued that diagnostic criteria are of less importance.
The Villefranche criteria evolved in 2017, keeping most of the same clinical criteria but with the addition of some further clinical criteria (Table 1).6 The accuracy of the 2017 criteria was assessed retrospectively in 50 patients with genetic confirmation of vEDS. Within this cohort, 32 were female with 24% having died at the time of the retrospective review. The mean age at death was 29 years. Forty percent of patients had had an acute arterial event (rupture/dissection) before the age of 40 years and 22% an unexplained sigmoid colon rupture. Two Villefranche major criteria were evident in 94% of patients, whereas only 28% of patients had all the major criteria. The frequencies of the other major and minor criteria are shown in Table 1.
There has been increasing diagnostic focus on both skin architecture and radiological investigation of large vessels. Ong et al used an ultrastructure scoring procedure following skin biopsy made up of abnormal fibroblast shape, presence of lysosomes in the fibroblast and abnormal basal lamina and showed it performed well with an AUC of 0.9.7 An older study which used immunofluorescence of cultured skin fibroblasts showed abnormal amounts of cytoplasm type III collagen.8 This result has been corroborated by abnormally low levels of serum procollagen type II aminopropeptide (P-III-NP), released during conversion of type III procollagen to collagen, in vEDS.9
Some groups have focused on haemodynamic and arterial wall properties. Arterial wall stress (both steady and pulsatile) in vEDS patients was found to be higher than in controls.10 Further, carotid intima-media thickness was 32% lower in patients with vEDS. An abnormally low intima-media thickness generates a higher wall stress which may increase the risk of arterial dissection and rupture in vEDS patients. Patients with vEDS have been shown to have a reduction in the relative increase in carotid pulse wave velocity between early and end systole, which could reflect a less adaptative arterial wall stiffening during the cardiac cycle.11 It was postulated that this may explain the higher susceptibility to arterial rupture in vEDS patients.
Novel imaging techniques have allowed for a detailed analysis of patients with vEDS. Using MRI of both the aorta and carotid arteries, Kerwin et al compared 17 patients with vEDS with eight age/sex-matched controls and analysed a number of artery-specific variables.12 They found that, in those patients with vEDS, there was a significant negative correlation (r=–0.82, p=0.02) between age-adjusted pulse propagation velocity and familial longevity, suggesting that elevated pulse propagation velocity (an indicator of vessel distensibility) may be a risk factor for complications of EDS IV. Ultrasound has also been used to examine the biomechanical properties of arteries in vEDS.13 Examination of the carotid artery has shown that common carotid artery distension and compliance tended to be lower in vEDS subjects.
Medical management
A key medical therapy in patients with vEDS is celiprolol (Table 2). Celiprolol is a β-blocker with a unique pharmacologic profile; it is a β1-adrenoceptor antagonist with partial β2 agonist activity.17 While it has antihypertensive and antianginal actions, reducing heart rate and pulsatile pressure, it lacks the commonly seen side effects of more mainstream β-blockers (namely bronchoconstriction, left ventricular function depression and peripheral vasoconstriction) due to the allied β2 agonist effect. This β2 agonist effect also influences vascular tone and directly affects smooth muscle and, as such, it is unique in being recognised as a therapeutic intervention for patients with vEDS. The reduction in heart rate and pulsatile pressure also reduces the mechanical stress on collagen fibres within the arterial wall.
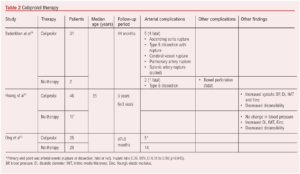
The Beta-Blockers in Ehlers-Danlos Syndrome (BBEST) study is a prospective, multicentre, randomised, open trial which reported in 2010.16 The study compared patients with vEDS randomised to either celiprolol or no treatment and the assessment of clinical events was blinded. Fifty-three patients were randomly assigned to celiprolol (25 patients) or control groups (28). The mean duration of follow-up was 47 (SD 5) months, with the trial stopped early for treatment benefit. The primary endpoints (arterial events – rupture or dissection, fatal or not) were reached by five (20%) in the celiprolol group and by 14 (50%) controls (hazard ratio 0.36; 95% CI 0.15 to 0.88; p=0.040). The beneficial results have been supported by more recent observational studies and, as such, celiprolol holds promise for the management of patients with vEDS.14
Surgical management
The fragile nature of the arterial system in patients with vEDS makes surgical management challenging. A study by Freeman et al reviewed their experience of managing vEDS in an era prior to the routine use of endovascular therapies.18 Within their series there were 22 spontaneous haemorrhages, 17 aneurysms and five arterial dissections. Eleven of the vascular complications were treated with non-operative management, eight with vessel ligation, 20 with arterial reconstruction, and two with endovascular therapy. Eighteen patients underwent diagnostic angiography, with three (22%) major complications and one death (5.6%). Twelve (30%) patients died from vascular complications of vEDS, seven of whom had previously been treated with arterial reconstruction. They concluded that vascular complications should be treated wherever possible with non-operative therapy and non-invasive imaging techniques. If required, operative intervention ideally should focus on vessel ligation rather than reconstruction.
Okada et al reported their experience of endovascular therapies for vEDS patients presenting with arterial complications.19 The interventions were predominantly for the development of false aneurysms, with the primary management being embolisation using a combination of coils and glue. While all procedures were ultimately successful, there were recognised complications both intra-procedurally and following the procedure both with access site issues and recurrence of the false aneurysm.
A more recent review of practice came from Oderich et al who reported their experience of managing 24 vEDS patients with vascular-related issues.20 In total, these 24 patients developed 132 vascular-specific complications (85 present before or at the time of the initial review and 47 occurring after the initial presentation). Fifteen patients underwent 30 operative interventions, of which 15 were reconstructive in nature. Three hospital deaths occurred due to uncontrollable haemorrhage. Procedure-related complications were high with one in three patients experiencing post-intervention bleeding and one in five patients requiring re-operation. The rate of late graft-related complications was also high, affecting 40% of arterial reconstructions and predominantly being due to anastomotic complications.
Natural history
This section focuses on the natural history of the condition with regard to vascular-specific complications as opposed to other complications including those affecting the gastrointestinal or genitourinary tracts. It is recognised that vEDS is associated with other non-vascular complications including gastrointestinal perforation (Table 3), complications around pregnancy and cardiac-related complications. The management of other allied health problems is also more complex in such patients.
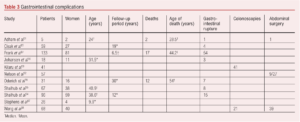
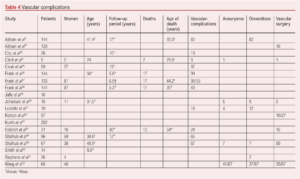
The natural history of vEDS with a focus on vascular complications shows that arterial complications are common, present as predominantly dissection or aneurysmal-related, and occur in a large proportion of patients (Table 4). Vascular complications can occur in any arterial bed, can develop at pace and the progression of asymptomatic aneurysmal disease as well as arterial dissections is uncertain. Life expectancy is shorter than that expected in patients without vEDS.
Discussion
vEDS is a rare condition and is associated with a significant risk of arterial complications. These provide significant challenges for vascular surgical teams to manage. The overall aim of this review was to highlight these complexities including offering a patient’s perspective on pathways of care (see below). There are a number of take-home messages from this review and the accompanying patient experience for vascular surgeons and other medical staff who will see patients with vEDS.
vEDS is caused by pathological alteration in the sequence variants of COL3A1, which codes for procollagen III. This leads to abnormalities in the quantity and quality of type III collagen. We have purposely not focused on the associated genetic defects associated with vEDS as we want this review to focus on clinically relevant issues. That said, the diagnosis of vEDS needs to be confirmed by either genetic analysis of the COL3A1 gene or by analysis of a patient’s collagen. vEDS is associated with a low intima-media thickness which, allied with high mechanical stress, leads to the high risk of arterial rupture and dissection. Close links with geneticists, dermatologists and other medical teams is essential in the longer-term management of such patients.
Within the UK, there are recognised centres of expertise. The EDS National Diagnostic Service is a highly specialised service commissioned by NHS England for individuals and families who are suspected to have complex EDS. These have been established since 2009 and are run out of the Sheffield Northern General Hospital and the Northwick Park & St Mark’s Hospitals in London. These services aim to provide an accurate diagnosis in suspected cases, they develop guidelines and pathways of care for the different EDS subtypes and provide information for patients and carers. They also help lead research on EDS. Their teams are multifaceted with a strong emphasis on genetics and counselling. Current diagnostic techniques include clinical review, skin biopsy and genetic blood testing. Imaging parameters may hold promise about predicting progression to a vascular event, but coalescence of expertise and patient numbers are probably required to achieve appropriate larger scale natural history studies.
The EDS National Diagnostic Service does not provide a pathway for vascular surgery intervention. These centres do not act as a referring centre for the vascular complications, and it is possible that a patient with vEDS may present to any emergency department in the UK and the patient perspective in this paper reflecting the challenges they face is powerful. Indeed, this paper provides a unique view in as much as we have included a patient perspective on the current acute management of patients with vEDS. While this is a single patient’s experience, it resonates with experiences of other patients and charities are actively engaging with numerous medical specialties (including emergency department staff) focusing on triage, the signs and symptoms of vascular emergencies and the need for a low threshold for imaging as patients can commonly present with relatively minor symptoms underlying a major arterial problem.
Currently there are no evidence-based guidelines for surveillance, and it is likely that an individualised approach is required. The lack of high-quality natural history data also make decision making regarding when to intervene in asymptomatic complications difficult. Again, more comprehensive natural history studies will help fill this gap in knowledge.
There is only one pharmacotherapy with a strong evidence base for reducing complication rates in patients with vEDS. The randomised controlled trial by Ong et al of the oral β-blocker celiprolol showed a significant reduction in arterial complications after a mean follow-up of 47 months (50% vs 20%).16 Celiprolol is a cardioselective β1 blocker with a β2 agonist vasodilatory effect. This results in a reduction in both heart rate and pulse pressure leading to a reduction in mechanical stress on the collagen fibres within the arterial wall. All patients should be considered for celiprolol therapy administered twice daily and titrated up to a maximum dose of 400 mg daily.
With the focus of this review being on human studies, we have not addressed the ongoing research being conducted in animal models which investigate new therapies.
The fragility of the arteries in this cohort of patients makes any arterial intervention complex and challenging. This is reflected in the outcomes from the three cohorts highlighted in this review. The lack of other large cohort studies and the larger number of case reports reflects the uncommon nature of the condition. The take-home message from the studies appears to be to treat any vascular complication non-operatively where possible, simplify the operative technique if intervention is required, endovascular intervention is appropriate and can be first line to arrest haemorrhage and that intervention can fail early and this needs to be recognised within the early post-intervention period. Patients with vEDS can develop arterial complications within any vessel and not just the aorta and, as such, endovascular intervention can provide less risk than open surgical management of difficult to access/manage vessels. Arterial access complications affecting either the femoral or brachial arteries can be easier to manage than more challenging management of the primary arterial complication. In the UK, such patients should be managed wherever possible within centres with the range of skills that may be required throughout the patient’s acute management.
The review has obvious limitations. The evidence base on vEDS is limited, which reflects the relatively rare nature of the condition, yet the overarching aim of this review is to draw attention and focus on the condition and to stimulate discussion within vascular surgical services and networks in the UK about how they would manage patients with vEDS and vascular complications.
Conclusion
vEDS is an uncommon condition often with a delayed diagnosis and life-threatening complications. Our understanding of vEDS is improving, but it is important that all vascular surgeons are aware of the condition, that it has a low threshold for investigation of minor symptoms and that they have access to the required facilities to treat such patients.


Article DOI:
Journal Reference:
J.Vasc.Soc.G.B.Irel. 2023;2(2):83-90
Publication date:
February 28, 2023
Author Affiliations:
1. Cambridge Medical School, University of Cambridge, UK
2. Leeds Vascular Institute, Leeds General Infirmary, Leeds, UK
Corresponding author:
Patrick Coughlin
Leeds Vascular Institute, Leeds General Infirmary, Great George Street, Leeds, LS1 3EX, UK
Email: [email protected]


")

")

